This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison
Gene Phylogeny
Phylogenies are constructed to indicate the relatedness of a set of organisms. In this case, the gene sequences are entered and their similarities used to determine which are most likely to have close common ancestors. Parsimony, or Occam's razor, is the idea that the simplest explanation used to get to the solution is usually the right one [1]. This is applied to phylogenetics by assuming that the gene sequences with the least number of changes are likely the ones that are most closely related through evolution.
Analysis
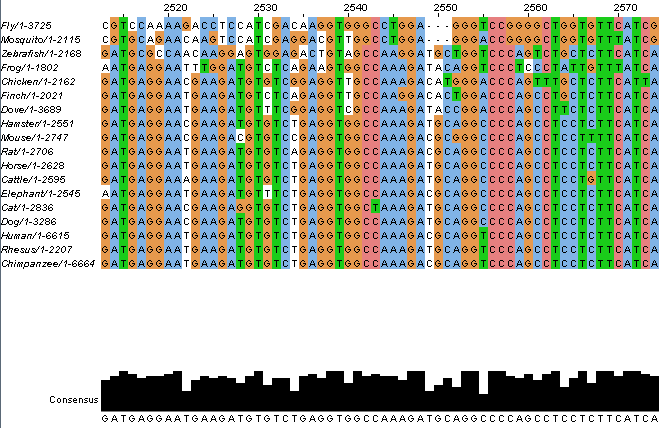
All gene sequences for the homologs listed above were used to construct a sequence alignment as well as several potential phylogenetic trees. ClustalOmega was used to perform the algorithms necessary for these constructs. Starting with the sequence alignment, all gene sequences had to be entered into a text file following a specific format so that ClustalOmega would interpret the data correctly. A sample document with the correct format is shown below.
This document can be pasted into ClustalOmega and the gene sequences will be aligned to the best of the program's ability. The output shows the aligned sequences with different coloring options such as hydrophobicity, percentage identity, and helix propensity among others, as well as the consensus at each position of the alignment.
One option within the sequence alignment window is to calculate a phylogenetic tree. The trees can either be Average Distance or Neighbor Joining trees, using Percent Identity or BLOSUM62 analysis methods. Percent Identity compares the sequences and looks at how similar they are compared to each other to determine the percent of bases that are identical between two strands [2]. In contrast, BLOSUM62 uses a distance matrix to score two aligned sequences based on the substitutions that occur between them . Those with the least substitutions have the lowest scores [3]. Each of these can then be used to build one of trees. Average Distance trees are structured so the sequences most similar to each other are organized with more recent common ancestors. The branch lengths are measured based on how many differences are found between the sequences, assuming that they diverged equally from the common ancestor [4]. Neighbor Joining trees begin with a single node and sequences are grouped together to generate the tree with the shortest branches. Branches are different lengths because it is not assumed that they have diverged equally from their common ancestor and therefore may have different mutation rates over time [5]. All four trees as they were constructed in ClustalOmega can be seen below.
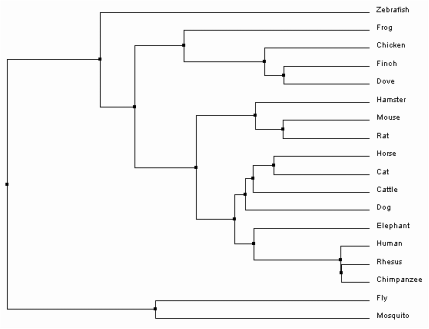
Average Distance Tree using Percent Identity
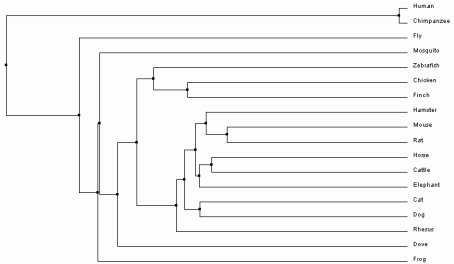
Average Distance Tree Using BLOSUM62
|
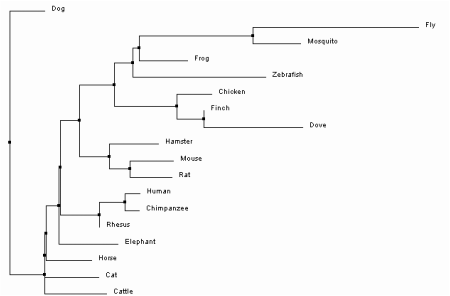
Neighbor Joining Tree using Percent Identity
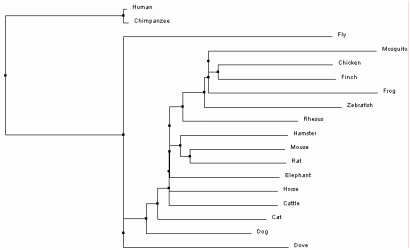
Neighbor Joining Tree using BLOSUM62
|
Discussion
Based on the trees above, it seems that the Average Distance tree using Percent Identity is the most plausible. The Neighbor Joining tree using Percent Identity places Canis lupus familiaris as the outgroup. This separates one member of the class Mammalia from the rest, and also shows divergence of the insects Drosophila melanogaster and Culex quinquefasciatus after birds and mammals. Likewise, both trees using BLOSUM62 have separated chimps and humans into the outgroup. Since they are also part of class Mammalia, it is more likely that they are related to other mammals such as Macaca mulatta. Only the Average Distance tree using Percent Identity puts the insects into the outgroup. It also shows humans and non-human primates as the most recently diverged organisms of those sampled, which has been confirmed through previous evolutionary studies [6].
References
ClustalOmega: http://www.ebi.ac.uk/Tools/msa/clustalo/
[1] Definition: Occam's Razor. (n.d.). Retrieved March 20, 2015, from http://www.merriam-webster.com/dictionary/occam's razor
[2] Tree Calculation. (n.d.). Retrieved March 21, 2015, from http://www.jalview.org/help/html/calculations/tree.html
[3] Eddy, S. (2004). Where did the BLOSUM62 alignment score matrix come from? Nature Biotechnology, 22, 1035-1036. Retrieved March 20, 2015, from http://www.nature.com/nbt/journal/v22/n8/full/nbt0804-1035.html
[4] Matsen IV, F., Gallagher, A., & McCoy, C. (2013). Minimizing the average distance to a closest leaf in a phylogenetic tree. Systematic Biology, 62(6), 824-824. Retrieved March 21, 2015, from http://connection.ebscohost.com/c/articles/95728315/minimizing-average-distance-closest-leaf-phylogenetic-tree
[5] Saitou, N., & Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol, 4(4), 406-425. Retrieved March 20, 2015, from http://mbe.oxfordjournals.org/content/4/4/406.short
[6] History of Life on Earth. (2014, October 1). Retrieved March 21, 2015, from http://www.bbc.co.uk/nature/history_of_the_earth
[1] Definition: Occam's Razor. (n.d.). Retrieved March 20, 2015, from http://www.merriam-webster.com/dictionary/occam's razor
[2] Tree Calculation. (n.d.). Retrieved March 21, 2015, from http://www.jalview.org/help/html/calculations/tree.html
[3] Eddy, S. (2004). Where did the BLOSUM62 alignment score matrix come from? Nature Biotechnology, 22, 1035-1036. Retrieved March 20, 2015, from http://www.nature.com/nbt/journal/v22/n8/full/nbt0804-1035.html
[4] Matsen IV, F., Gallagher, A., & McCoy, C. (2013). Minimizing the average distance to a closest leaf in a phylogenetic tree. Systematic Biology, 62(6), 824-824. Retrieved March 21, 2015, from http://connection.ebscohost.com/c/articles/95728315/minimizing-average-distance-closest-leaf-phylogenetic-tree
[5] Saitou, N., & Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol, 4(4), 406-425. Retrieved March 20, 2015, from http://mbe.oxfordjournals.org/content/4/4/406.short
[6] History of Life on Earth. (2014, October 1). Retrieved March 21, 2015, from http://www.bbc.co.uk/nature/history_of_the_earth
